Double digestion sangat umum ditemui dalam konstrak DNA. Bisa mudah kita bayangkan kita mau potong insert atau vektor, enzim restriksi ini adalah guntingnya. Untuk bekerja optimal (100%), maka diperlukan kondisi tertentu yang disediakan oleh buffer tertentu pula. Double digestion akan jadi mudah kalau buffer dari 2 enzim yang digunakan adalah sama. Nah, kalau beda, ini kudu mikir.
Secara umum, ada 3 jenis buffer universal yaitu L buffer, M buffer, dan H buffer. Untungnya, Toyobo menyediakan tabel ini, yaitu daftar enzim dan buffer mana yang digunakan untuk menghasilkan kerja efisiensi 100%.
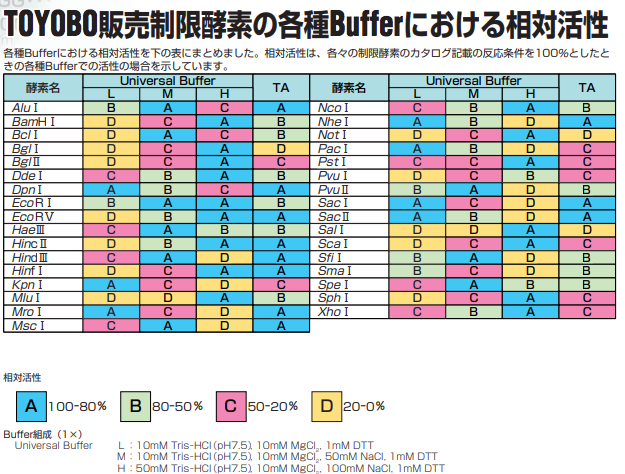
Perhatikan, ada kode ABCD yang menunjukkan efisiensi pemotongan pada buffer tertentu (LMH). Dan biasanya buffer yang disediakan oleh pabrik yaitu 10X sehingga komponennya cukup dikali 10 dari yang tertulis di atas
L bufer misalnya menjadi:
- 100 mM Tris-HCl (pH7.5)
- 100 mM MgCl2
- 10 mM Dithiothreitol (DTT)
H buffer menjadi:
- 500 mM Tris-HCl (pH 7.5)
- 100 mM MgCl2
- 1000 mM NaCl
- 10 mM DTT
Sekarang kita masuk ke kasus nyata, dimana saya mau potong DNA pakai ApaI dan BamHI. Di label, ApaI pakai L buffer, sedangkan BamHI pakai H buffer.
Cara potongnya:
- Selalu mulai dari yang komponen buffernya L, terus M, dan H. Sehingga kali ini saya potong pertama kali adalah dengan ApaI. Potong selama 1 jam, kemudian dilanjut potong pakai BamHI.
- Coba perhatikan komponen buffernya. L dan H buffer berbeda pada konsentrasi Tris-HCl dan NaCl-nya, sedangkan lainnya sama. Sehingga pada pemotongan kedua yang pakai BamHI, saya ga perlu pakai H buffer, tapi cukup adjust dari komponen yang kurang dengan stok Tris-HCl dan NaCl konsentrasi tinggi yang ada di lab.
Adjust yang saya lakukan saat BamHI:
- Volume total = 10 uL
- L-Buffer 1 uL
- Tris HCl = 0.5 uL (stock 1 M)
- NaCl = 0.4 uL (stock 2.5 M)
Resume komposisi reaksi
Komposisi reaksi yang saya lakukan untuk ApaI:
- DNA = 5.5 uL (stock 116.5 ng)
- Nuclease free water = 3.2 uL
- Enzyme ApaI = 0.3 uL
- L buffer = 1 uL
37 derajat 1 jam
- Lanjut BamHI dengan adjust di atas.
37 derajat 1 jam
================
Plasmid contain 6 T:
- DNA 0.8 uL
- Nuclease free water = 7.9 uL
- Enzyme ApaI = 0.3 uL
- L buffer = 1 uL
Star activity
Tau kan ini? Ini artinya pemotongan tidak spesifik yang bisa dikarenakan oleh beberapa hal. Misal untuk BamHI diperngaruhi oleh:
- konsentrasi garam yang rendah
- kelebihan enzim
- konsentrasi tinggi gliserol
- adanya Mn2+